Sickle Cell Explained
Sickle Cell Disorders (SCD) are inherited chronic illnesses that affect all ethnic groups. They occur more frequently in people of African, Caribbean, Middle Eastern, Indian and Mediterranean descent. Sickle Cell anaemia is the most common of these inherited conditions. SCD affects many parts of the body and is associated with severe painful crises.
People with Sickle Cell anaemia have a type of haemoglobin (called haemoglobin S (HbS) or Sickle haemoglobin) which differs from usual haemoglobin (haemoglobin A or HbA). Haemoglobin is the substance in our red blood cells that gives blood its red appearance. The function of haemoglobin is to transport oxygen from the lungs to the rest of the body.
The transfer of oxygen takes place in the narrow blood vessels called capillaries. These may be no more than the width of a red blood cell. In people with usual haemoglobin A, the red blood cells remain round and flexible to enable them to squeeze through the capillaries. However in people with Sickle haemoglobin, the haemoglobin forms into long rigid chains.
These chains distort the cell membrane into odd shapes - often like the shape of the old-fashioned farming implement called a Sickle. Such Sickle-shaped cells are not as flexible and easily become stuck in the narrow blood vessels. When this happens, that part of the body becomes deprived of oxygen. The result is mild, moderate or excruciating pain in the part of the body affected, with the possibility of permanent damage to the tissue.
The current treatments for Sickle Cell anaemia are painkillers for the crises, and blood transfusions for particular types of crises where the bone marrow production of new blood cells collapses. The most recent promising treatment involves a drug called hydroxyurea, which has been shown to reduce the incidence of crises, though at the cost of some side effects.
The aim is therefore to try to prevent the complications that can arise with Sickle Cell anaemia. Penicillin is given daily to try to ward off infections which, especially for the first seven years of life, could be life threatening for children.
A full and up-to-date set of vaccinations is also vital for people with Sickle Cell. Folic acid supplements are given to promote the production of red blood cells. Certain factors have been identified as more likely to precipitate a painful sickle cell crisis. These include infections, cold/damp conditions, dehydration, strenuous exertion, stress, sudden changes in temperature, alcohol, smoking and anaesthetics.
Advice to sufferers on preventing crises therefore includes keeping warm, eating healthily, taking moderate exercise, taking plenty of fluids, and keeping up to date with medications and vaccinations.
Sickle Cell Carriers
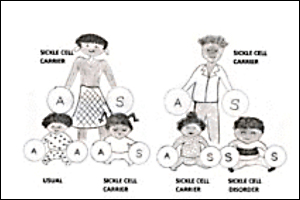
A Sickle Cell carrier is someone who carries a gene associated with Sickle Cell, but who, except in rare exceptional circumstances, is usually perfectly healthy themselves. There are about 250,000 Sickle Cell carriers (people with Sickle Cell trait) in the UK with around 15,000 people living with a sickle cell disorder.
If both biological parents are Sickle Cell carriers, then in each pregnancy there is a one in four chance that they will have a child with Sickle Cell anaemia; a one in four chance they will have a child with usual haemoglobin; and a one in two chance that they will have a child who is a Sickle Cell carrier.